Plotting
pharokka_plotter.py
Pharokka v1.3.0 implements pharokka_plotter.py, which creates a simple circular genome plot using pyCirclize with output in PNG format. All CDS are coloured according to their PHROG functional group.
It is reasonably customisable and is designed for single input phage contigs. If an input FASTA with multiple contigs is entered, it will only plot the first contig. It requires the input FASTA, Pharokka output directory, and the -p or --prefix value used with Pharokka if specified.
You can run pharokka_plotter.py in the following way (most basic command below).
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory
This will create pharokka_plot.png as an output file plot of your phage in the pharokka output directory.
A prefix is not required for pharokka by default. If you used a prefix to create your pharokka output, please specify it:
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory -p my_prefix
If you want to give your plot a title (e.g. phage lambda):
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory -t 'phage lambda'
To add italics (especially for genus or species names), encase them with ${ }$ as follows:
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory -t '${Escherichia}$ phage lambda'
If you want to make the title bigger:
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory -t '${Escherichia}$ phage lambda' --title_size 30
By default all CDSs that are not hypothetical or unknown are labelled. If you want to reduce this (if the plot is overcrowded for example), use --annotations to chose the proportion of annotations that you want labelled (descending based on size).
For example, the following command will label half the annotations
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory --annotations 0.5
You can also use this to remove all CDS labels by specifying --annotations 0
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory --annotations 0
By default hypothetical or unknown genes are not labelled. If you want to label them use --label_hypotheticals:
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory --label_hypotheticals
If you want the axis intervals to be changed (e.g. 10kbp here):
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory -t '${Escherichia}$ phage lambda' --title_size 30 --interval 10000
By default all axis labels longer than 20 characters are truncated. To change this number to whatever you want, use --truncate.
For example this will truncate all labels to 15 characters:
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory --truncate 15
If you want to make the CDS labels bigger (defaults to 8):
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory --label_size 10
If you want to change the plot resolution (default 600 dpi)
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory -dpi 600
If you want to specify pharokka gff and genbank files instead of a directory, use --gff and genbank. Note that this will save pharokka_plot.png in the working directory.
pharokka_plotter.py -i input.fasta -n pharokka_plot --gff pharokka.gff --genbank pharokka.gbk
By default, all tRNAs, tmRNAs and CRISPRs will labelled. If you want to turn this off, please use --remove_other_features_labels
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory --remove_other_features_labels
If you want to specify specific labels, you can do this by including a list of locus_tags (from the .gff, .gbk or the summary output files) as a text file with one ID per line and specify this file with --label_ids:
e.g. if you make a text file called labels.txt
EHOONAYF_CDS_0007
EHOONAYF_CDS_0009
EHOONAYF_CDS_0023
pharokka_plotter.py -i input.fasta -n pharokka_plot -o pharokka_output_directory --label_ids labels.txt
usage: pharokka_plotter.py [-h] -i INFILE [-n PLOT_NAME] [-o OUTDIR] [--gff GFF] [--genbank GENBANK] [-p PREFIX] [-t PLOT_TITLE] [-f]
[--label_hypotheticals] [--remove_other_features_labels] [--title_size TITLE_SIZE] [--label_size LABEL_SIZE]
[--interval INTERVAL] [--truncate TRUNCATE] [--dpi DPI] [--annotations ANNOTATIONS] [--label_ids LABEL_IDS]
pharokka_plotter.py: pharokka plotting function
options:
-h, --help show this help message and exit
-i INFILE, --infile INFILE
Input genome file in FASTA format.
-n PLOT_NAME, --plot_name PLOT_NAME
Output plot file name. ".png" suffix will be added to this automatically.
Will be output in the pharokka output directory if -o is specified, or in the working directory if --gff andf --genbank are specified.
-o OUTDIR, --outdir OUTDIR
Pharokka output directory.
--gff GFF Pharokka gff.
--genbank GENBANK Pharokka genbank.
-p PREFIX, --prefix PREFIX
Prefix used to create pharokka output. Will default to pharokka.
-t PLOT_TITLE, --plot_title PLOT_TITLE
Plot name.
-f, --force Overwrites the output plot file.
--label_hypotheticals
Flag to label hypothetical or unknown proteins. By default these are not labelled.
--remove_other_features_labels
Flag to remove labels for tRNA/tmRNA/CRISPRs. By default these are labelled.
They will still be plotted in black.
--title_size TITLE_SIZE
Controls title size. Must be an integer. Defaults to 20.
--label_size LABEL_SIZE
Controls annotation label size. Must be an integer. Defaults to 8.
--interval INTERVAL Axis tick interval. Must be an integer. Must be an integer. Defaults to 5000.
--truncate TRUNCATE Number of characters to include in annoation labels before truncation with ellipsis.
Must be an integer. Defaults to 20.
--dpi DPI Resultion (dots per inch). Must be an integer. Defaults to 600.
--annotations ANNOTATIONS
Controls the proporition of annotations labelled. Must be a number between 0 and 1 inclusive.
0 = no annotations, 0.5 = half of the annotations, 1 = all annotations.
Defaults to 1. Chosen in order of CDS size.
--label_ids LABEL_IDS
Text file with list of CDS IDs (from gff file) that are guaranteed to be labelled.
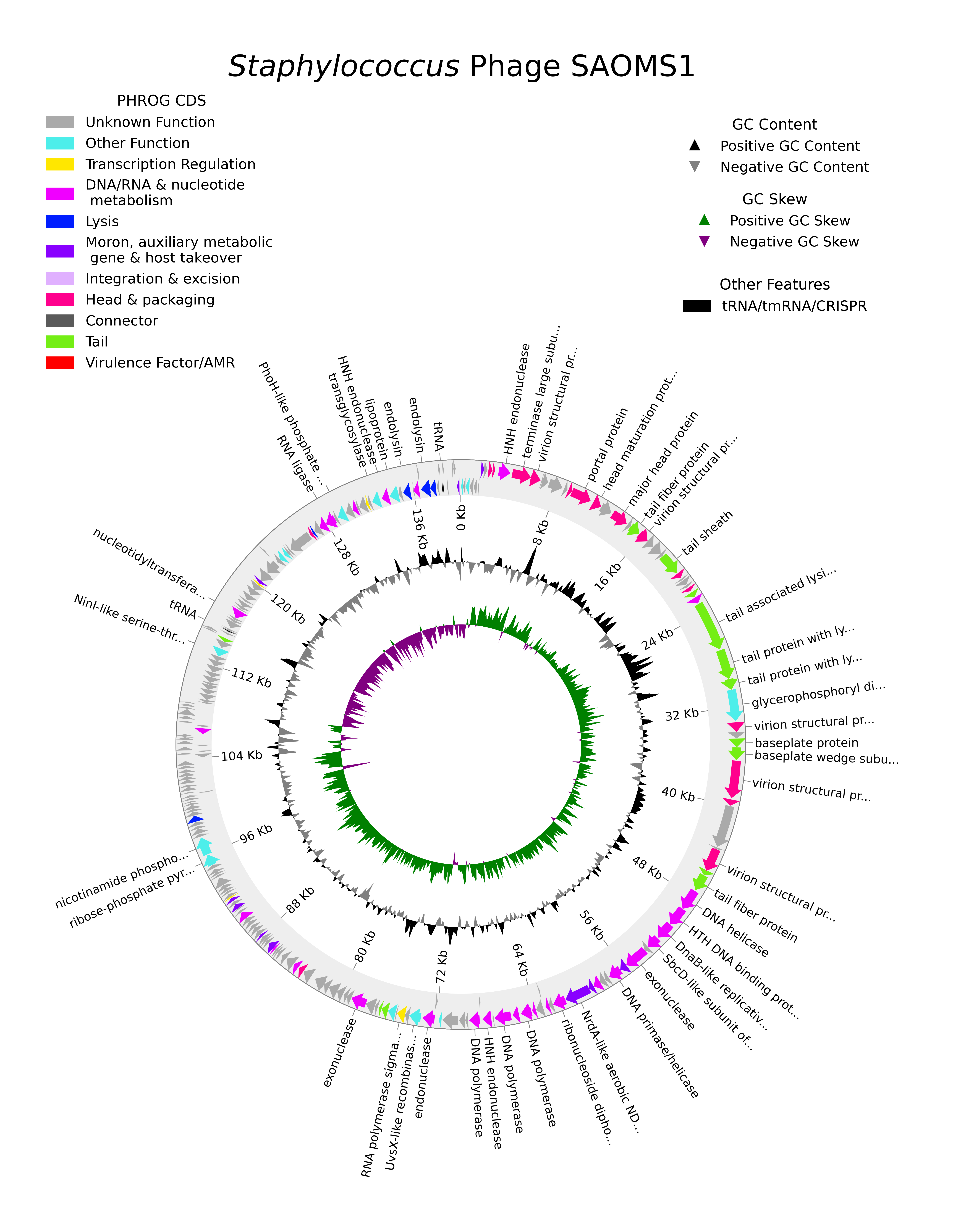
SAOMS1 phage (GenBank: MW460250.1) was isolated and sequenced by: Yerushalmy, O., Alkalay-Oren, S., Coppenhagen-Glazer, S. and Hazan, R. from the Institute of Dental Sciences and School of Dental Medicine, Hebrew University, Israel.
pharokka_multiplotter.py
After receiving a number of requests, pharokka v1.7.0 implements pharokka_multiplotter.py that can be used to plot multiple contigs.
It requires the pharokka output Genbank file (here, pharokka.gbk). It will save plots for each contig in the output directory (here pharokka_plots_output_directory).
e.g.
pharokka_multiplotter.py -g pharokka.gbk -o pharokka_plots_output_directory
The rest of the parameters are the same as pharokka_plotter.py
usage: pharokka_multiplotter.py [-h] -g GENBANK -o OUTDIR [-f] [--label_hypotheticals] [--remove_other_features_labels]
[--title_size TITLE_SIZE] [--label_size LABEL_SIZE] [--interval INTERVAL] [--truncate TRUNCATE] [--dpi DPI]
[--annotations ANNOTATIONS] [-t PLOT_TITLE] [--label_ids LABEL_IDS]
pharokka_multiplotter.py: pharokka plotting function for muliple phages
options:
-h, --help show this help message and exit
-g GENBANK, --genbank GENBANK
Input genbank file from Pharokka.
-o OUTDIR, --outdir OUTDIR
Pharokka output directory.
-f, --force Overwrites the output plot file.
--label_hypotheticals
Flag to label hypothetical or unknown proteins. By default these are not labelled.
--remove_other_features_labels
Flag to remove labels for tRNA/tmRNA/CRISPRs. By default these are labelled.
They will still be plotted in black.
--title_size TITLE_SIZE
Controls title size. Must be an integer. Defaults to 20.
--label_size LABEL_SIZE
Controls annotation label size. Must be an integer. Defaults to 8.
--interval INTERVAL Axis tick interval. Must be an integer. Must be an integer. Defaults to 5000.
--truncate TRUNCATE Number of characters to include in annoation labels before truncation with ellipsis.
Must be an integer. Defaults to 20.
--dpi DPI Resultion (dots per inch). Must be an integer. Defaults to 600.
--annotations ANNOTATIONS
Controls the proporition of annotations labelled. Must be a number between 0 and 1 inclusive.
0 = no annotations, 0.5 = half of the annotations, 1 = all annotations.
Defaults to 1. Chosen in order of CDS size.
-t PLOT_TITLE, --plot_title PLOT_TITLE
Plot name.
--label_ids LABEL_IDS
Text file with list of CDS IDs (from gff file) that are guaranteed to be labelled.